The computer simulation of complex systems often requires approaches that cover very different length-scales as in describing a photo-active chromophore embedded in a large protein or a solvated molecule. To this aim, one often employs hybrid multiscale methods where a more accurate description is adopted for the active site and a coarser one for the rest of the system. We outline below two recent contributions of our group in the field of multiscale methods for the study of excited-state properties of embedded systems.
Wave function embedding for excited states
Embedding potentials are frequently used to account for the effect of an environment on the electronic structure of molecules in larger systems, including their excited states. If the excitation is accompanied by significant rearrangements in the electron density of the embedded molecule, the environment is expected to strongly respond to this perturbation. We have recently developed a simple, practical scheme to accurately compute the excitation energies of a molecule embedded in a responsive environment within a wave-function-in-density-functional-theory framework. Through the use of state-specific potentials, the approach accounts for the mutual polarization of the fragments upon excitation of the active species.
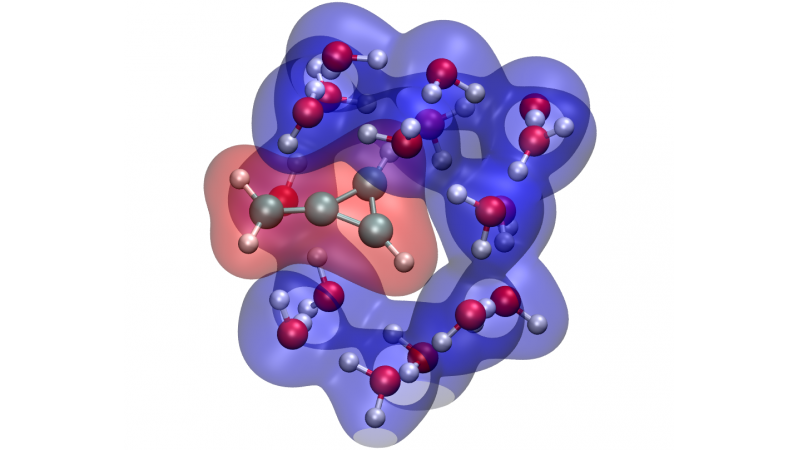{kind=link}
- C. Daday, C. Konig, J. Neugebauer, and C. Filippi, Wavefunction-in-density functional theory embedding for excited states: Which wavefunctions, which densities?, ChemPhysChem 15, 3205 (2014).
Quantum Monte Carlo in polarizable force fields for excited states
We have recently developed a quantum mechanics/molecular mechanics approach that combines accurate quantum Monte Carlo methods with the reaction field of classical polarizable dipoles (QMC/MMpol). A different set of optimal dipoles are self-consistently generated at the variational Monte Carlo level in equilibrium with the states involved in the electronic transition and then used to include electrostatic and inductive environmental effects in the description of the photoexcited molecule.
{kind=link}
R. Guareschi, H. Zulfikri, C. Daday, F. M. Floris, C. Amovilli, B. Mennucci, and C. Filippi, Introducing QMC/MMpol: Quantum Monte Carlo in polarizable force fields for excited states, J. Chem. Theory Comput. 12, 1674 (2016).