Dynamic Solids at near-critical conditions with Machine-Learning Force Fields
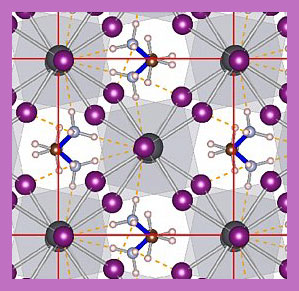
![]() Lattice dynamics at the atomic scale is often well described by phonons in the harmonic approximation. In recent years, however, anharmonicity has been shown to play an important role in, for instance, the ultra-low thermal conductivity of thermoelectric materials with high figures-of-merit. While ab-initio molecular-dynamics (MD) captures the anharmonicity correctly, it is computationally orders of magnitude too expensive to describe the effects of phonon scattering related to the "rattling" and "flipping" of atoms and molecules. We develop novel methods that can account for these effects based upon an efficient and robust on-the-fly Machine-Learning Force-Field (MLFF) method. This is a very recent development that allows us to automatically construct smooth models of the potential energy surface computed by density functional theory. The new methods will be used to simulate the lattice dynamics in "Dynamic Solids" and gives access to the nanosecond time- and tens of nanometer length-scales required in MD simulations. The resulting method will have near first-principles accuracy and capture both the anharmonic potential as well as the rapid formation and breaking of weak bonds.
Lattice dynamics at the atomic scale is often well described by phonons in the harmonic approximation. In recent years, however, anharmonicity has been shown to play an important role in, for instance, the ultra-low thermal conductivity of thermoelectric materials with high figures-of-merit. While ab-initio molecular-dynamics (MD) captures the anharmonicity correctly, it is computationally orders of magnitude too expensive to describe the effects of phonon scattering related to the "rattling" and "flipping" of atoms and molecules. We develop novel methods that can account for these effects based upon an efficient and robust on-the-fly Machine-Learning Force-Field (MLFF) method. This is a very recent development that allows us to automatically construct smooth models of the potential energy surface computed by density functional theory. The new methods will be used to simulate the lattice dynamics in "Dynamic Solids" and gives access to the nanosecond time- and tens of nanometer length-scales required in MD simulations. The resulting method will have near first-principles accuracy and capture both the anharmonic potential as well as the rapid formation and breaking of weak bonds.
The halide-perovskites are a prime example of materials where both effects play an important role. These are promising materials for new solar and thermoelectric technologies. As well as simulating their lattice thermal conductivity and NMR dipolar coupling, we will attempt to resolve fundamental structure-property relationss. We want to bring the MLFF method out of the development lab and into the workshop that is the interdisciplinary Dutch materials research community.
Funding by the University of Twente in the form of a tenure-track.
Dr. Menno Bokdam (PI)
Dr. Jonathan Lahnsteiner (Post-Doc)
Recording of my presentation at the Molecular Simulation with Machine Learning On-line workshop this summer. It presents the hybrid perovskites as a prime example of "Dynamic Solids" and gives overview of the research and developments we have made in the last 3 years.
Dynamics in perovskite photovoltaics
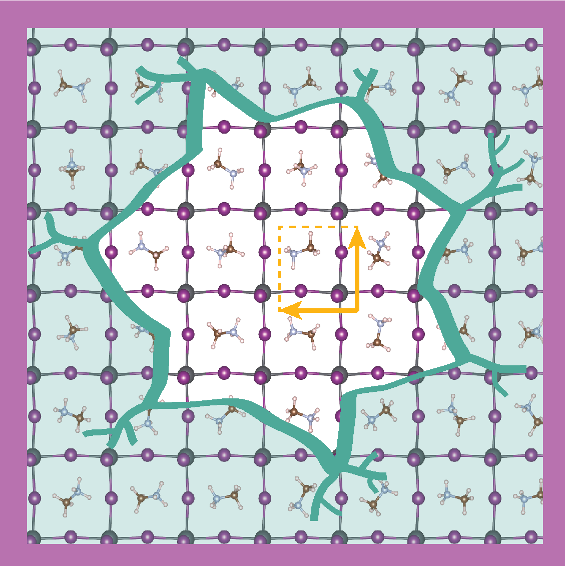
![]() A rat race for the highest efficiency perovskite solar cell has emerged out of the initial report that organo-metal halide perovskites can function as a photovoltaic dielectric. The last three years have seen an astonishing increase from approximately 10% in 2012 to more than 20% efficiency in 2015. This material is made up of elements abundantly found in nature, has a simple production procedure and can therefore be used to produce cheap solar cells. However, the main issue preventing perovskites solar cells from going to market is their lack of stability. Where silicon solar cells have live times of about 20 years, a perovskite solar cells typically breaks down after several days. The bad material strength, however, is at the same time accompanied with the presence of polar phonons and freely rotatable methylammonium (MA) molecules in the material. The vibrations of the ionic lattice and the intrinsic dipole moment of the MA molecule can screen slowly oscillating electric fields. In a foregoing study we have started to understand the role of these polar phonons in photo excited state of the perovskite. In an attempt to stabilize the perovskite under ambient conditions researchers have placed a single layer of the 2D-material hexagonal Boron-Nitride on top. This material is transparent for light, but does not allow humidity to pass trough. How this interface affects the molecular ordering is however unknown and will influence the solar cells efficiency. We propose to focus on the orientation of the MA molecules in the bulk material as well as at the interface with a 2D material and determine under which conditions they show a long range ordered behaviour. The orientation of the MA molecules cannot be uniquely obtained from experiment, however large scale molecular dynamics have shown to do just that. With these calculations we would like to find an answers to the superseding question: Do the methylammonium molecules in the record breaking MAPbI3 perovskite make an essential contribution to its solar cell success, and if so, how?
A rat race for the highest efficiency perovskite solar cell has emerged out of the initial report that organo-metal halide perovskites can function as a photovoltaic dielectric. The last three years have seen an astonishing increase from approximately 10% in 2012 to more than 20% efficiency in 2015. This material is made up of elements abundantly found in nature, has a simple production procedure and can therefore be used to produce cheap solar cells. However, the main issue preventing perovskites solar cells from going to market is their lack of stability. Where silicon solar cells have live times of about 20 years, a perovskite solar cells typically breaks down after several days. The bad material strength, however, is at the same time accompanied with the presence of polar phonons and freely rotatable methylammonium (MA) molecules in the material. The vibrations of the ionic lattice and the intrinsic dipole moment of the MA molecule can screen slowly oscillating electric fields. In a foregoing study we have started to understand the role of these polar phonons in photo excited state of the perovskite. In an attempt to stabilize the perovskite under ambient conditions researchers have placed a single layer of the 2D-material hexagonal Boron-Nitride on top. This material is transparent for light, but does not allow humidity to pass trough. How this interface affects the molecular ordering is however unknown and will influence the solar cells efficiency. We propose to focus on the orientation of the MA molecules in the bulk material as well as at the interface with a 2D material and determine under which conditions they show a long range ordered behaviour. The orientation of the MA molecules cannot be uniquely obtained from experiment, however large scale molecular dynamics have shown to do just that. With these calculations we would like to find an answers to the superseding question: Do the methylammonium molecules in the record breaking MAPbI3 perovskite make an essential contribution to its solar cell success, and if so, how?
Funding by the Austrian Science Fund (FWF): P 30316-N27
Run-time: 01.07.2017 - 30.06.2020
Dr. Menno Bokdam (PI)
Jonathan Lahnsteiner MSc (PhD-student)
Status: Finished