Assessing density functionals using many body theory for hybrid perovskites
One of the most important issues in the modelling of materials is
the choice of an appropriate density functional. Many researchers
employ functionals commonly used in their field or they have some
sort of chemical intuition, why one density functional should be
preferred over another one. This is an ill-advised strategy. In this
paper, we present a concise approach to select the best functional
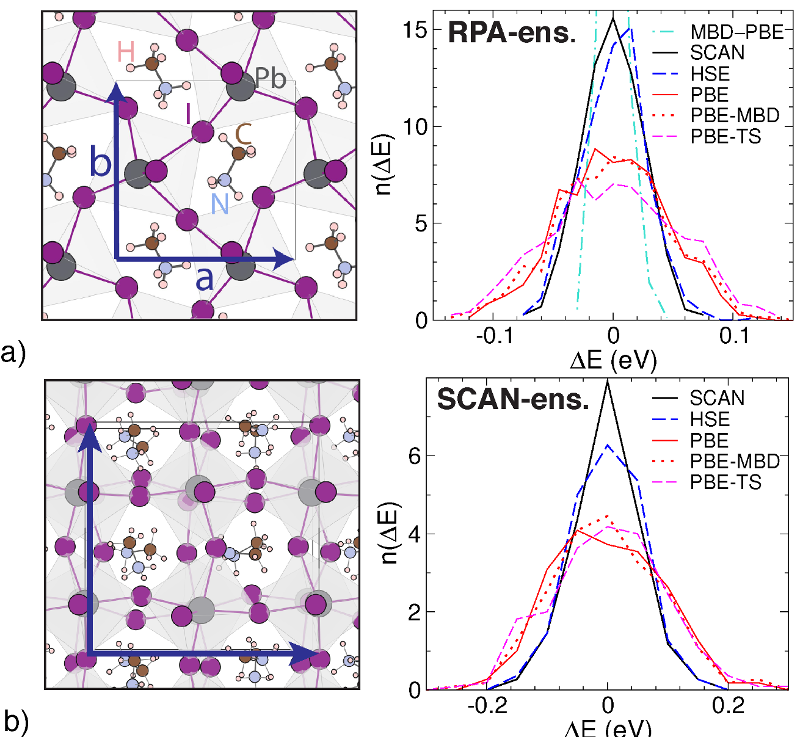
for structure prediction of a particular materials system. We use the
random phase approximation (RPA), which is placed one step above
hybrid functionals on the metaphorical Jacob's Ladder towards the
exact total energy. A new implementation of RPA-forces in VASP
(by Ramberger and Kresse, 2016) allows us to perform molecular
dynamics at the RPA level, something that seemed impossible just
a few years ago. A finite temperature ensemble of realistic crystal
structures and the associated energies are calculated. Comparing
these energies to the ones obtained with commonly used density
functionals allows to rank them based on their accuracy.
To verify this new approach, we study an exciting novel solar cell
material: MAPbI3. Its structure is a particularly hard nut to crack
for DFT. This is due to the large dynamical degree of freedom of
the Methylammonium molecules and the interplay of van der Waals
forces and cage instabilities in the perovskite structure.
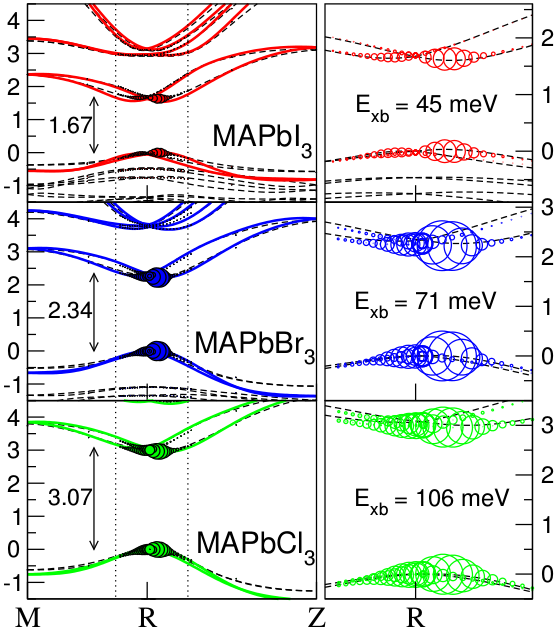
Role of Polar Phonons in the Photo Excited State of Metal Halide Perovskites
We have calculated the band structure and exciton binding energies
of the most studied ABX3 hybrid perovskites. We have incorporated
many body effects on the DFT calculated electronic structure in
the GW0 approximation and consecutively solved the Bethe-Salpeter
equation (BSE). Convergence of the red-shift of the optical band
gap requires the use of very dense k-point grids. We have therefore
implemented a modelBSE routine in VASP, where a model screening
function, fitted to W0, is used.
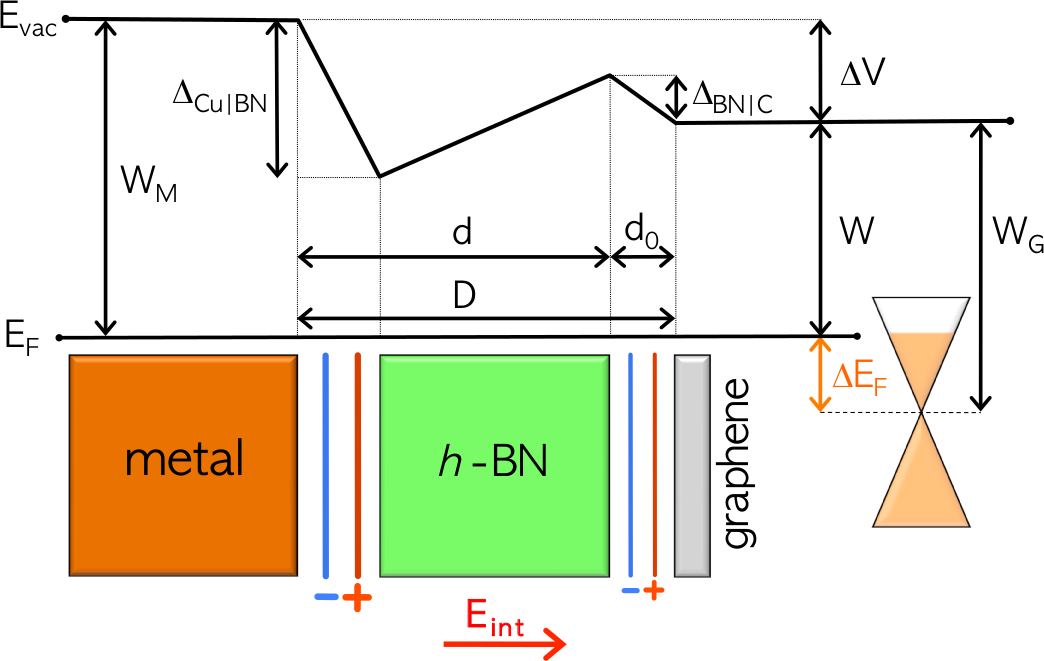
Electrostatic Doping of Graphene through Ultrathin Hexagonal Boron Nitride Films
A model that accurately describes the doping
level in graphene on a h-BN covered metal surface is presented. The model is
based on the electrostatical description of a simple planar capacitor.
Interface bonding effects are included as localized potential steps and
are obtained independently by first principles calculations. The doping
level can be tuned by an external electric field and the metal contact
results in a non-trivial intrinsic doping.
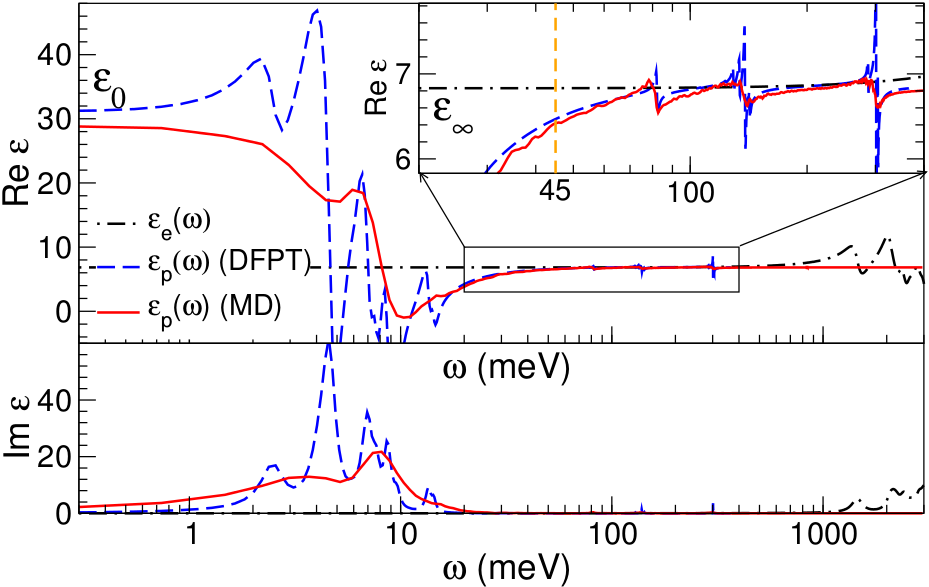
Role of Polar Phonons in the Photo Excited State of Metal Halide Perovskites
We have solved an open issue in this field, whether ionic movement
can screen an e-h pair and thereby effectively lower the exciton binding
energy. For this purpose we have calculated the room temperature
dielectric function from molecular dynamics. By following the
fluctuations of the total dipole moment in time, the polarizability
can be calculated in linear response.
Band gaps in incommensurable graphene on hexagonal boron nitride
Early DFT calculations of commensurate graphene on h-BN showed a
small induced band gap of approximately 40 meV. Low temperature STM images
later showed that the graphene|h-BN is in reality incommensurate as
indicated by the formation of large moir ́e patterns. We have shown
that this does not have to mean that the induced band gap disappears
and that a band gap of similar order can form. Since the required
super cells are so large, Kohn-Sham DFT is not a viable option.
We have therefore constructed a tight-binding model based on GW
calculations for commensurate structures.
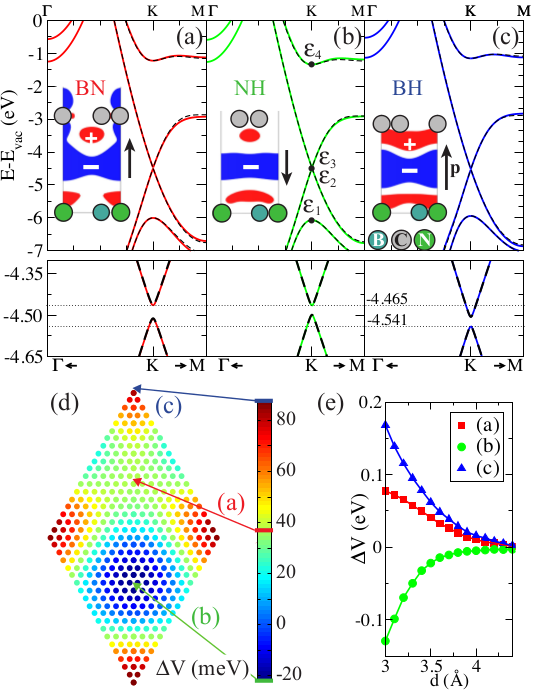
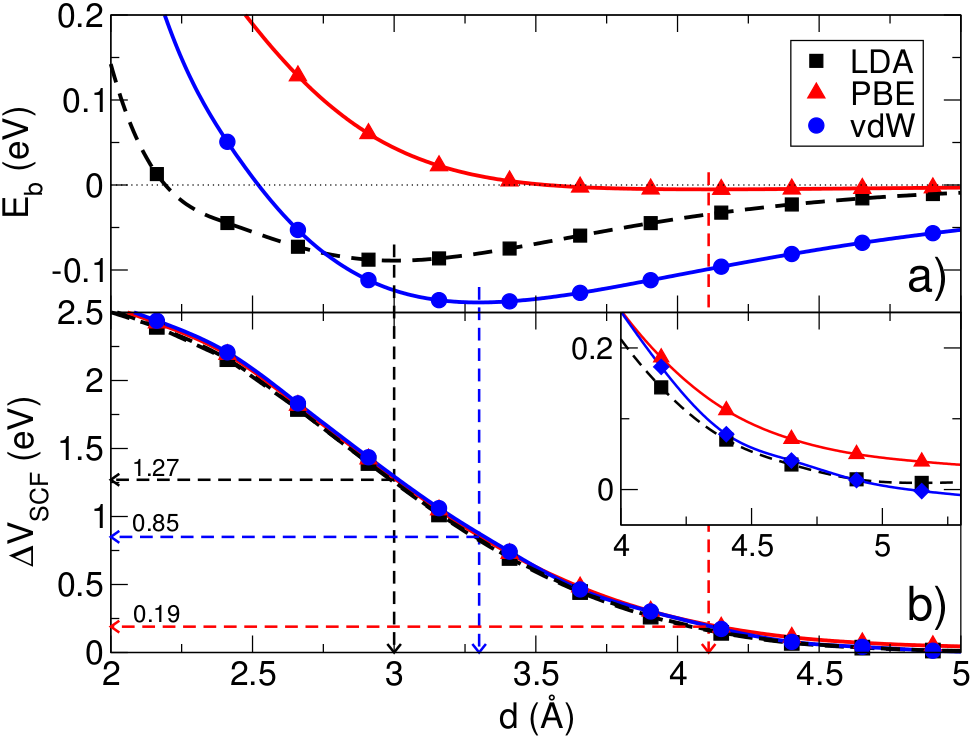
Large potential steps at weakly interacting metal-insulator interfaces
At metal-insulator interfaces large potential steps (1 eV) can be
formed even though the interaction is of a weak van der Waals
type. We have used the interface between a metal and h-BN
(M|BN) as a archetypical example to study the underlying physical
mechanisms. As shown in the top figure, DFT is unable to predict
the equilibrium binding distance. However, the induced potential
step as a function of distance does not depend on the XC-functional.
We have approximated the M|BN wavefunction by constructing an
anti-symmetric product of the individual M and BN wavefunctions
in a self-adapted version of VASP. The resulting system is a good
description of the self-consistently calculated M|BN system. This
proofs directly that the interface dipole is formed by Pauli exchange
repulsion.
Phase Transitions of Hybrid Perovskites Simulated by Machine-Learning Force Fields Trained on the Fly with Bayesian Inference
Realistic finite temperature simulations of matter are a formidable challenge for first principles methods. Long simulation times and large length scales are required, demanding years of computing time. Here we present an on-the-fly machine learning scheme that generates force fields automatically during molecular dynamics simulations. This opens up the required time and length scales, while retaining the distinctive chemical precision of first principles methods and minimizing the need for human intervention. The method is widely applicable to multielement complex systems. We demonstrate its predictive power on the entropy driven phase transitions of hybrid perovskites, which have never been accurately described in simulations. Using machine learned potentials, isothermal-isobaric simulations give direct insight into the underlying microscopic mechanisms. Finally, we relate the phase transition temperatures of different perovskites to the radii of the involved species, and we determine the order of the transitions in Landau theory.
Peer-reviewed publications
2024
Jansen T., Jannis D., Bouwmeester R.L., Zhang Z., Bokdam M., Koster G., Gauquelin N., Verbeeck J., and Brinkman A.,"Phase transitions of LaMnO3 and SrRuO3 from DFT+U based machine learning force fields simulations", Phys. Rev. Materials 8, 125002, DOI: 10.1103/PhysRevMaterials.8.125002
Lahnsteiner J., Rang M. and Bokdam M., "Tuning Einstein Oscillator Frequencies of Cation Rattlers: A Molecular Dynamics Study of the Lattice Thermal Conductivity of CsPbBr3", J. Phys. Chem. C 128 (3), 1341-1349, DOI: 10.1021/acs.jpcc.3c06590
2023
Jansen T., Brocks G., and Bokdam M.,"Phase transitions of LaMnO3 and SrRuO3 from DFT+U based machine learning force fields simulations", J. Phys. Rev. B 108, 235122, DOI: 10.1103/PhysRevB.108.235122
Kim HJ., Ahn J., Voronina N., Yaqoob N., Bokdam M., Jeong J., Park JH., Chung KY., Kaghazchi P., Myung ST., "Impact of water on structure stabilization in layered manganese-oxide for high-voltage zinc storage in non-aqueous electrolyte: Experimental and theoretical aspects", Energy Storage Materials, 63, 103028, DOI: 10.1016/j.ensm.2023.103028
Biega R., Bokdam M., Herrmann K., Mohanraj J., Skrybeck D., Thelakkat M., Retsch M., Leppert L. , "Dynamic Distortions of Quasi-2D Ruddlesden–Popper Perovskites at Elevated Temperatures: Influence on Thermal and Electronic Properties", J. Phys. Chem. C, 127, 19, 9183–9195, DOI: 10.1021/acs.jpcc.3c01634
Fykouras K., Lahnsteiner J., Leupold N, Tinnemans P, Moos R, Panzer F, de Wijs G.A., Bokdam M.,Grueninger H. and Kentgens A.P.M. , "Disorder to order: how halide mixing in MAPbI3−xBrx perovskites restricts MA dynamics", J. Mater. Chem. A, 11, 4587-4597 , DOI: 10.1039/D2TA09069D
2022
Lahnsteiner J., Bokdam M., " Anharmonic lattice dynamics in large thermodynamic ensembles with machine-learning force fields: CsPbBr3 a phonon liquid with Cs rattlers", Phys. Rev. B 105, 024302, DOI: 10.1103/PhysRevB.105.024302
2021
Bokdam M., Lahnsteiner J., Sarma D.D., "Exploring Librational Pathways with on-the-Fly Machine-Learning Force Fields: Methylammonium Molecules in MAPbX3 (X = I, Br, Cl) Perovskites", J. Phys. Chem C, 125, 21077-21086, DOI: 10.1021/acs.jpcc.1c06835
Varrassi L., Liu P., Ergonec Z.,, Bokdam M., Kresse G. and Franchini C., "Optical properies of transition metal oxide perovskites by the Bethe-Salpeter equation", Phys. Rev. Materials 5, 074601, DOI: 10.1103/PhysRevMaterials.5.074601
Grueninger H., Bokdam M.; Leupold N.; Tinnemans P., Moos R., De Wijs G., Panzer F. and Kentgens A., "Microscopic (dis)order and dynamics of cations in mixed FA/MA lead halide perovskites", J. Phys. Chem. C, 125, 1742-1753, DOI: 10.1021/acs.jpcc.0c10042
2019
Lahnsteiner J., Jinnouchi R., Bokdam M., "Long-range order imposed by short-range interactions in methylammonium-lead iodide MAPbI3: Comparing point-dipole models to machine-learning force fields", Phys. Rev. B. 100, 094106, DOI: 10.1103/PhysRevB.100.094106
Lahnsteiner J., Kresse G., Heijnen J., Bokdam M., "The Finite Temperature Structure of the MAPbI3 Perovskite:
Comparing density functional approximations and force fields to experiment", Phys. Rev. Mat., 2, 073604 (2018) DOI: 10.1103/PhysRevMaterials.2.073604
2017
Bokdam M., Lahnsteiner J., Ramberger B., Schäfer T., Kresse G. "Assessing density functionals using many
body theory for hybrid perovskites", Physical Review Letters, 119, 145501, (2017), DOI: 10.1103/PhysRevLett.119.145501
Hu S., Gao H., Qi Y., Tao Y., Li Y., Reimers J.R., Bokdam M., Franchini C., Di Sante D., Stroppa A., Ren
W. "Dipole Order in Halide Perovskites: Polarization and Rashba Band Splittings", Journal of Physical
Chemistry C, 121, 23045-23054 (2017) DOI: 10.1021/acs.jpcc.7b05929
Govinda S., Kore B.P., Bokdam M., Mahale P., Kumar A., Pal S., Bhattacharyya B., Lahnsteiner J., Kresse
G., Franchini C., Pandey A., Sarma D.D. "Behavior of Methylammonium Dipoles in MAPbX(3) (X = Br
and I)", Journal of Physical Chemistry Letters, 8, 4113-4121 (2017) DOI: 10.1021/acs.jpclett.7b01740
2016
Lahnsteiner J., Kresse G., Kumar A., Sarma D.D., Franchini C., Bokdam M. "Room-temperature dynamic
correlation between methylammonium molecules in lead-iodine based perovskites: An ab initio molecular
dynamics perspective", Physical Review B, 94, 214114 (2016) DOI: 10.1103/PhysRevB.94.214114
Bokdam M., Sander T.,Stroppa A., Picozzi S., Sarma DD., Franchini C., Kresse G., "Role of Polar Phonons
in the Photo Excited State of Metal Halide Perovskites", Scientific Reports, 6, 28618, (2016), DOI:
10.1038/srep28618
Amlaki T., Bokdam M., Kelly PJ., "Z 2 invariance of Germanene on MoS 2 from first principles", Physical
Review Letters, 116, 256805, (2016), DOI: 10.1103/PhysRevLett.116.256805
2014
Stroppa A., Di Sante D., Barone P., Bokdam M., Kresse G., Franchini C., Whangbo MH., Picozzi S.,
"Tunable ferroelectric polarization and its interplay with spin-orbit coupling in tin iodide perovskites",
Nature Communications, 5, 5900, (2014), DOI: 10.1038/ncomms6900
Bokdam M., Brocks G., Kelly PJ., "Large potential steps at weakly interacting metal-insulator interfaces",
Physical Review B (Rap. Comm.), 90, 201411, (2014), DOI: 10.1103/PhysRevB.90.201411
Bokdam M., Brocks G., Katsnelson MI., Kelly PJ., "Schottky barriers at hexagonal boron nitride/metal in-
terfaces: A first-principles study ", Physical Review B, 90, 085415, (2014), DOI: 10.1103/PhysRevB.90.085415
Bokdam M., Amlaki T., Brocks G., Kelly PJ., "Band gaps in incommensurable graphene on hexagonal boron
nitride", Physical Review B (Rap. Comm.), 89, 201404, (2014), DOI: 10.1103/PhysRevB.89.201404
2013
Bokdam M., Khomyakov PA., Brocks G., Kelly PJ., "Field effect doping of graphene in metal|dielectric
|graphene heterostructures: A model based upon first-principles calculations", Physical Review B, 87,
075414, (2013), DOI: 10.1103/PhysRevB.87.075414
2012
Brocks G., Cakir D., Bokdam M., de Jong, MP., Fahlman M., "Charge equilibration and potential steps in
organic semiconductor multilayers", Organic Electronics, 13, 1793, (2012), DOI:
10.1016/j.orgel.2012.05.041
Cakir D., Bokdam M.; de Jong, MP., Fahlman M., Brocks G, "Modeling charge transfer at organic donor-
acceptor semiconductor interfaces, Applied Physics Letters", 100, 203302, (2012), DOI: 10.1063/1.4717985
2011
Bokdam M., Khomyakov PA., Brocks G., Zhong ZC., Kelly PJ., "Electrostatic Doping of Graphene through
Ultrathin Hexagonal Boron Nitride Films", Nano Letters, 11, 4631, (2011), DOI: 10.1021/nl202131q
Bokdam M., Cakir D.; Brocks G., "Fermi level pinning by integer charge transfer at electrode-organic
semiconductor interfaces", Applied Physics Letters, 98, 113303, (2011), DOI: 10.1063/1.3565963
Phd thesis
15 November 2013
Bokdam M., "Charge transfer and redistribution at interfaces between metals and 2D materials", University of Twente Library, Enschede, The Netherlands, DOWNLOAD