Phase transitions using on-the-fly MLFF mentioned in scientific background to the Nobel prize in Physics '24
Wednesday, 9. October 2024

I congratulate John J. Hopfield and Geoffrey E. Hinton on winning the 2024 Nobel Prize in Physics for their foundational discoveries in machine learning using artificial neural networks (ANNs). According to the official Nobel Prize announcement, Hopfield and Hinton’s pioneering work laid the groundwork for the ANN architectures that are transforming everything from scientific research to everyday technologies. Their achievements have helped revolutionize fields such as physics, chemistry, and even healthcare, by enabling computers to learn, adapt, and solve complex tasks much like biological systems. The pair’s contributions—especially in the development of recurrent and feedforward networks—have unlocked the power of artificial intelligence and made deep learning an integral part of modern science.
Their pioneering work has also been instrumental in developing Machine Learning Force Fields (MLFF) to study phases of complex materials with an accuracy comparable to ab initio quantum-mechanical models and a speedup by a factor as large as 1000! I am very honoured that the Physics Nobel prize comittee has mentioned our work on simulating phase transitions using MLFFs as powerful new tools in physics. See reference [40] in the 'Scientific Background to the Nobel Prize in Physics 2024'.
Also in the prize annoucement the possibilities to find new solar cell materials were mentioned:
What a nice surpise!
Menno Bokdam
FAIR Data Fund project: Creating a cockpit-like overview of large materials simulation databases – 4TU.ResearchData
Tuesday, 26. September 2023

Menno Bokdam gave an interview to 4TU.researchdata about the outcomes of the Fair Data Fund Project.
The link to the full interview is here.
University Teaching Qualification Award Ceremony
Tuesday, 15. June 2023

At the University of Twente a lot of care and attention is spend on the study programmes. Therfore, all teachers at the university are expected to continuously evaluate and improve their courses. The university teaching qualification (UTQ) is a programme intended to assist new university staff members in this process. Today several teachers of the Science and Technology faculty received a certificate signifying the successfull completion of the UTQ. Among them was Menno Bokdam who received the UTQ from the director of the Applied Physics programme Sissi de Beer.
FAIR datafund arwarded
Monday, 13. February 2023
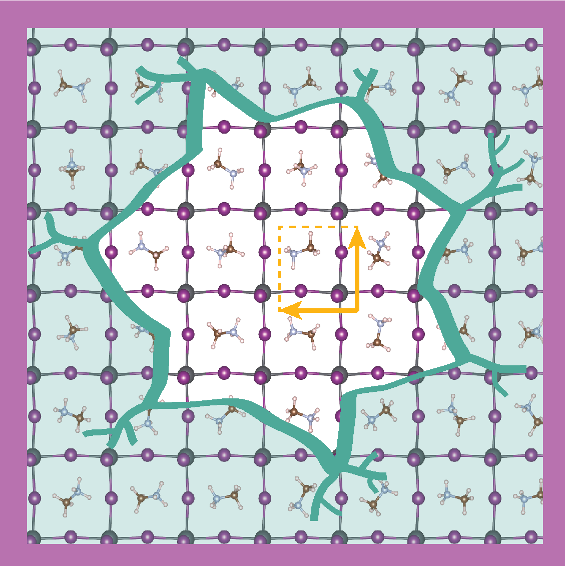
Our propsal for the FAIR Data Fund 2022 Autumn Call was funded to the value of € 3500,-. Congratulations to Thijmen Kuipers, it was his BSc project that lead to this proposal. The application was reviewed independently by three reviewers. The funds will be used to make the software developed by Thijmen avaialble to the scientific community. About the project: This project places the scientist in the ‘cockpit’ of a database of materials simulated at the atomic scale. These databases are so richly filled with information that it is difficult to assess its contents. We generate an overview of the most important physical properties in the database and their distributions. It enables a quick executive decision; to use, extend or discard this database for new simulations.
New computing cluster up and running.
Monday, 21. December 2020
Our new extension to the compute cluster is now fully operational. This extension was made possible by pooling resources of Prof. Paul Kelly, Dr. Linn Leppert and Dr. Menno Bokdam. It consists out of 20 nodes, each containing 2 Intel Xeon Gold 6242 2.8G, 16C/32T processors supported by 8x32Gb memory. Fast communication between the nodes (essential for efficient parallel computing) is made by Melanox Infiniband Adapters. Special thanks goes to Frederik Reenders and Dr. Jonathan Lahnsteiner for tuning the new system's performance.



Move to the University of Twente
Tuesday, 2. June 2020
After "six wonderful and scientifically fruitful years" at the University of Vienna, Menno Bokdam moved to the University of Twente were he will start in a new Tenure Track position.
![]() ---------->>>>>
---------->>>>>
![]()
Effective atomic interactions in complex materials picked up by on-the-fly machine-learning
Friday, 21. June 2019
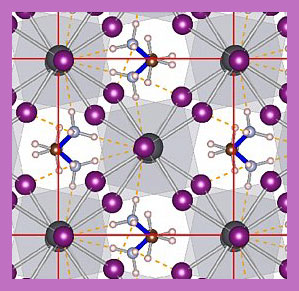
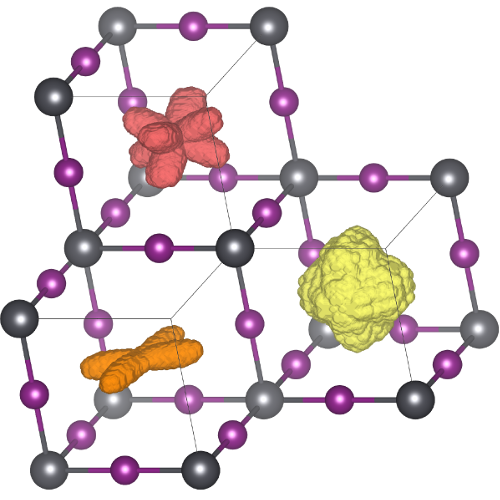 At the atomic scale materials can show a rich palette of dynamic behaviour, that directly affects the physical properties of these materials. For many years, it has been a dream to describe these dynamics in complex materials at various temperatures using computer simulations. Physicists of the Computational Materials Physics Group at the University of Vienna have developed an on-the-fly machine-learning method that enables such calculations through direct integration into the quantum mechanics based Vienna Ab-initio Simulation Package (VASP). The versatility of the method is demonstrated by presenting new microscopic insight on the phase transitions of hybrid perovskites. These materials attract enormous scientific interest, because of their potential in solar energy harvesting and other applications. The results of this work recently appeared in the journal Physical Review Letters.
At the atomic scale materials can show a rich palette of dynamic behaviour, that directly affects the physical properties of these materials. For many years, it has been a dream to describe these dynamics in complex materials at various temperatures using computer simulations. Physicists of the Computational Materials Physics Group at the University of Vienna have developed an on-the-fly machine-learning method that enables such calculations through direct integration into the quantum mechanics based Vienna Ab-initio Simulation Package (VASP). The versatility of the method is demonstrated by presenting new microscopic insight on the phase transitions of hybrid perovskites. These materials attract enormous scientific interest, because of their potential in solar energy harvesting and other applications. The results of this work recently appeared in the journal Physical Review Letters.
At room temperature, all materials are constantly moving at the atomic scale. Even solid rock consists of atoms that swing around. The physical properties of materials are directly linked to the arrangement of atoms in the, so called, crystal lattice. Depending on the temperature or pressure this arrangement can change thereby affecting the materials properties. One can think of diamond, which is transparent and hard because of the periodic arrangement of carbon atoms in the diamond crystal. The same atoms, arranged differently, results in black, brittle graphite. It was already possible to accurately calculate the coordinates of the atoms in simple materials at different temperatures with quantum mechanical molecular dynamics (MD) simulations. However, such calculations are computationally expensive and restrict practical applications to a couple of hundreds of atoms and limited simulation time. Physicists from the Computational Materials Physics group at the University of Vienna have developed a new approach that overcomes these limitations and makes simulations of complex materials for future energy applications possible. This is achieved by improving existing machine learning methods and, most importantly, by integrating these methods directly into the Vienna Ab-initio Simulation Package (VASP). In the new approach, the "machine" can pick up, on its own, the essential ingredients for a simpler model description of the interacting atoms during MD simulations. Already after calculation a few hundreds of time-steps the machine can predict accurately enough the positions of the atoms in the consecutive time-step. The machine is also able to make an estimate of its accuracy for the consecutive steps. If the error is too high, the machine switches gears and performs the very accurate, but expensive, MD calculations. The more simulation time passes, the more the machine learns and the more precise it becomes. In this way, fewer and fewer MD calculations are required, which eventually leads to the situation where all time-steps are made by the machine. Moreover, the on-the-fly self-learning ability reduces the need for human intervention required by other existing machine-learning methods.
To demonstrate the power of this new method, the researchers have applied it to study the transitions between different atomic structures of the MAPbI3 perovskite upon changing the temperature. This material is very popular because of its potential as a new cheap solar cell component. It is made of organic molecules that can rapidly flip around, separated on a lattice composed of lead and iodide atoms. Depending on the temperature three different crystal phases are formed. The atomic mechanisms near the transition temperature are very difficult to be determined by experiment, and MD simulations would require years of compute time even on a modern super-computing system. After learning, the machine can predict the phase transition temperatures and lattice constants of this material with unprecedented precision. The developed method is general and applicable to many other future materials science problems and will become available for researchers word-wide in the upcoming version of VASP.
This project received funding by the Austrian Science Fund (FWF): P 30316-N27.
Best poster prize at COMDI18
Friday, 14. September 2018
At the "International Workshop on Computational Design and Discovery of Novel Materials" in Lausanne, Switzerland, Menno Bokdam was one of the winners of the Best Poster Award.

External website: COMDI18 - Posters
Launch of the new website.
Saturday, 23. June 2018
The "Dynamics in perovskite photovoltaics" project has now its own website. The old website will remain online, but will no longer be updated.
Our paper "Assessing Density Functionals Using Many Body Theory for Hybrid Perovskites" recently appeared in Physical Review Letters.
Thursday, 30. November 2017
One of the most important issues in the modelling of materials is the choice of an appropriate density functional. Many researchers employ functionals commonly used in their field or they have some sort of chemical intuition, why one density functional should be preferred over another one. This is an ill-advised strategy and a better approach is necessary. In this paper, we present a concise approach to select the best functional for a particular materials system using a state of the art method beyond density functional theory (DFT). We use the random phase approximation (RPA), which is placed one step above hybrid functionals on the metaphorical 'Jacob's Ladder' towards the exact total energy. This method has an accuracy unattained by any other computational method for solids. A breakthrough in the analytical formulation of forces in the RPA earlier this year makes this novel approach possible. An efficient implementation in the VASP code allows to perform molecular dynamics at the RPA level, something that seemed impossible just a few years ago. A finite temperature ensemble of realistic crystal structures and the associated energies are calculated. Comparing these energies to the ones obtained with commonly used density functionals allows to rank them based on their accuracy.
To verify this new approach, we study one of the most exciting novel light-harvesting materials: MAPbI3. Its structure is a particularly hard nut to crack for DFT. This is due to the large dynamical degree of freedom of the Methyl-ammonium molecules and the interplay of van der Waals forces and cage instabilities in the perovskite structure. A surprising outcome is that the SCAN functional outperforms a computationally much more costly hybrid functional. Furthermore, with SCAN we are for the first time able to show the nature of the dynamically stable tetragonal phase of MAPbI3 observed at room temperature.
Menno Bokdam, Jonathan Lahnsteiner, Benjamin Ramberger, Tobias Schaefer and Georg Kresse
Phys. Rev. Lett. 119, 145501 - Published 6 October 2017
Journal: https://doi.org/10.1103/PhysRevLett.119.145501
Open acces: https://arxiv.org/abs/1708.06821
Start project: "Dynamics in perovskite photovoltaics"
Saturday, 1. July 2017
A rat race for the highest efficiency perovskite solar cell has emerged out of the initial report that organo-metal halide perovskites can function as a photovoltaic dielectric. The last three years have seen an astonishing increase from approximately 10% in 2012 to more than 20% efficiency in 2015. This material is made up of elements abundantly found in nature, has a simple production procedure and can therefore be used to produce cheap solar cells. However, the main issue preventing perovskites solar cells from going to market is their lack of stability. Where silicon solar cells have live times of about 20 years, a perovskite solar cells typically breaks down after several days. The bad material strength, however, is at the same time accompanied with the presence of polar phonons and freely rotatable methylammonium (MA) molecules in the material. The vibrations of the ionic lattice and the intrinsic dipole moment of the MA molecule can screen slowly oscillating electric fields. In a foregoing study we have started to understand the role of these polar phonons in photo excited state of the perovskite. In an attempt to stabilize the perovskite under ambient conditions researchers have placed a single layer of the 2D-material hexagonal Boron-Nitride on top. This material is transparent for light, but does not allow humidity to pass trough. How this interface affects the molecular ordering is however unknown and will influence the solar cells efficiency. We propose to focus on the orientation of the MA molecules in the bulk material as well as at the interface with a 2D material and determine under which conditions they show a long range ordered behaviour. The orientation of the MA molecules cannot be uniquely obtained from experiment, however large scale molecular dynamics have shown to do just that. With these calculations we would like to find an answers to the superseding question: Do the methylammonium molecules in the record breaking MAPbI3 perovskite make an essential contribution to its solar cell success, and if so, how?
Funding by the Austrian Science Fund (FWF): P 30316-N27
Run-time: 01.07.2017 - 30.06.2020
Dr. Menno Bokdam (PI)
Jonathan Lahnsteiner MSc (PhD-student)